




金刚烷具有三维笼状碳骨架, 结构紧密, 具有很好的热稳定性, 是一类重要的有机化合物, 可广泛用于药物、航空航天、功能高分子、含能材料等领域[1]。若在金刚烷的环上引入一些高能或氧化基团, 如硝基、硝酰氧基等, 得到的硝基和硝酰氧基金刚烷具有较高的密度、爆速、爆压及能量[2-5]。自1980年Sollott等[6-7]以金刚烷为原料合成出1, 3, 5, 7-四硝基金刚烷以来, 人们已相继合成了多种硝基和硝酰氧基金刚烷, 其中包括:1, 3, 5, 7-四硝酰氧基金刚烷[8], 2, 2, 6, 6-四硝基金刚烷[9-10], 2, 2, 4, 4-四硝基金刚烷[11-12], 2, 2, 4, 4, 6, 6-六硝基金刚烷[13-14], 1, 2, 2-三硝基金刚烷[15]和4, 4, 8, 8-四硝基金刚烷-2, 6-二醇二硝酸酯[16]。此外, 用一个或多个氮原子替换金刚烷环上碳原子可以提高密度、爆速、爆压及其能量[17]。因此, 硝基和硝酰氧基氮杂金刚烷类化合物是一类具有较好应用前景的含能材料。目前关于硝基和硝酰氧基氮杂金刚烷的合成报道较少[18], 仅有3, 5, 7-三硝基-1 -氮杂金刚烷[19]和2, 4, 10-三硝基-2, 4, 10-三氮杂金刚烷[20]。
本研究以一氮杂金刚烷为母体, 在其骨架上引入两个硝酰氧基和一个硝基, 合成了一种新的氮杂金刚烷化合物—2-硝基-2-氮杂金刚烷-4, 8-二醇二硝酸酯, 表征了其结构和热性能。研究了硝化反应因素对2-硝基-2-氮杂金刚烷-4, 8-二醇二硝酸酯收率的影响, 确定了最佳的反应条件, 并预估了目标化合物的爆轰性能。
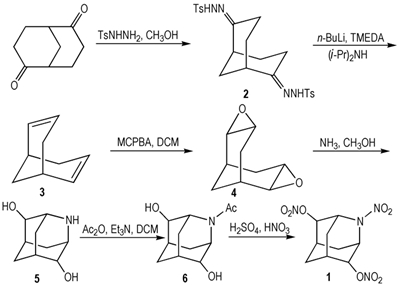
2 实验部分 2.1 合成路线合成路线见Scheme 1。

|
Scheme 1 |
仪器:Nicolet傅里叶变换红外光谱仪(美国Thermofisher公司); Bruker Avance-Ⅲ DRX 500 MHz核磁共振仪(德国Bruker公司); Vario EL-Ⅲ型元素分析仪(德国Elemetar公司); WRS-1B数字熔点仪(上海申光仪器仪表有限公司); Finnigan TSQ Quantumultra AM型质谱仪(美国Thermal公司); SDT Q600 DSC-TGA同步热分析仪(美国TA仪器公司)。
试剂:双环[3.3.1]壬烷-2, 6-二酮(自制[21]); 氨气(南京上元工业气体厂); 其它试剂均为分析纯, 使用前未经处理。
2.3 实验过程 2.3.1 双环[3.3.1]壬烷-2, 6-二烯(3)的合成双环[3.3.1]壬烷-2, 6-二烯采用两步法(即腙化、消除)合成[22], 具体步骤如下:
腙化:将2.25 g(14.82 mmol)双环[3.3.1]壬烷-2, 6-二酮溶于60 mL甲醇中, 加入6.6 g(35.5 mmol)对甲苯磺酰肼, 加热回流4 h后, 冷却, 抽滤得浅黄色固体, 再用少量乙醇洗涤, 真空干燥, 得6.75 g(13.81 mmol)白色固体2, 收率93%。
消除:将15.6 mL(15.6 mmol)i-Pr2NH、57 mL(493 mmol)四甲基乙二胺(TMEDA)加入到250 mL三口烧瓶中, 0 ℃下磁力搅拌, 再在氮气保护下慢慢滴入1.6 mol·L-1的n-BuLi/正己烷溶液45 mL(72 mmol), 将6.75 g(13.81 mmol)化合物2慢慢加入到上述体系, 加料过程持续约15 min, 加料过程中有大量气泡产生; 加料完毕后, 再常温搅拌15 h。反应结束后, 0 ℃下缓慢滴加50 mL水淬灭反应, 分液, 有机相再分别用10%的稀盐酸100 mL、饱和碳酸氢钠溶液80 mL、饱和食盐水80 mL洗涤, 洗涤后的有机相用无水硫酸钠干燥, 过滤, 40 ℃下减压蒸馏得1.16 g(9.71 mmol)浅黄色油状液体3, 收率70%。
1H NMR(CDCl3, 500 MHz) δ:5.79~5.71(m, 2H), 5.66(ddd, J1=1.20 Hz, J2=4.8 Hz, J3=9.95 Hz, 2H), 2.50~2.43(m, 2H), 2.23(ddd, J1=2.1 Hz, J2=3.60 Hz, J3=18.55 Hz, 2H), 1.86(dd, J1=4.70 Hz, J2=17.70 Hz, 2H), 1.70(t, J=2.90 Hz, 2H); 13C NMR(CDCl3, 126 MHz) δ:131.88, 125.15, 30.86, 28.68, 28.36。
2.3.2 2, 3;6, 7-二环氧双环[3.3.1]壬烷(4)的合成在250 mL单口瓶中加入5.76 g(85%, 28.41 mmol)间氯过氧苯甲酸(MCPBA)、60 mL二氯甲烷, 0 ℃下搅拌10 min后, 向体系缓慢滴入1.42 g(11.80 mmol)化合物3的二氯甲烷溶液60 mL, 滴加完毕后, 常温搅拌24 h。反应结束后, 慢慢滴入10%的亚硫酸氢钠水溶液40 mL, 搅拌至水相不再使淀粉-K I试纸变蓝。分液, 有机相再用5%的碳酸氢钠水溶液80 mL洗涤, 收集有机相, 水相继续用二氯甲烷萃取两次, 合并有机相, 用无水硫酸钠干燥, 过滤, 减压蒸馏得白色黏稠液体。中性氧化铝柱层析, 石油醚/乙醚(V:V=5:1)洗脱, 得1.25 g(8.20 mmol)白色固体4, 收率70%, m.p.171.6~173.2 ℃(文献值[23]:m.p.173~174 ℃)。
1H NMR(CDCl3, 500 MHz) δ:3.17~3.06(m, 2H), 2.97~2.86(m, 2H), 2.16(dd, J1=6.85 Hz, J2=15.75 Hz, 2H), 2.10~2.00(m, 2H), 1.88(dd, J1=4.70 Hz, J2=15.75 Hz, 2H), 1.42(t, J=2.95 Hz, 2H); 13C NMR(CDCl3, 126 MHz) δ:56.58, 50.15, 26.53, 24.41, 18.63;IR(KBr, ν/cm-1):2913, 1446(C—H), 1267, 825, 780(C—O—C); Anal.calcd for C9H12O2:C 71.03, H 7.95;found:C 71.09, H 7.97。
2.3.3 2-氮杂金刚烷-4, 8-二醇(5)的合成将0.8 g(5.26 mmol)化合物4、5.5 mL甲醇依次加入到35 mL厚壁耐压瓶中, 再加入6 mL饱和的氨气甲醇溶液(饱和的氨气甲醇溶液制备:向0 ℃下的甲醇缓慢通入氨气1 h), 密封后, 120 ℃下加热18 h。反应过程中有白色固体析出, 反应结束后, 冷却, 抽滤得0.78 g白色固体。再用乙醇重结晶, 得0.7 g(4.13 mmol)白色晶体5, 收率79%, m.p.322.4~323.9 ℃(文献值[23]:m.p.321 ℃)。
1H NMR(CD3OD, 500 MHz) δ:3.86(t, J=3.15 Hz, 2H), 2.86~2.81(m, 2H), 2.26(dt, J1=2.65 Hz, J2=13.0 Hz, 2H), 1.93~1.87(m, 2H), 1.84(t, J=2.96 Hz, 2H), 1.65(dt, J1=3.35 Hz, J2=13.05 Hz, 2H); 13C NMR(CD3OD, 126 MHz) δ:72.70, 51.53, 34.21, 31.04, 24.97;IR(KBr, ν/cm-1):3291, 3256(O—H, N—H), 2919, 1468(C—H), 1109, 1093(C—OH), 1061, 1049(C—N); Anal.calcd for C9H15NO2:C 63.88, H 8.93, N 8.28;found:C 63.91, H 8.89, N 8.31;ESI-MS(m/z):170 [M+H]+。
2.3.4 2-乙酰基-2-氮杂金刚烷-4, 8-二醇(6)的合成将1 g(5.90 mmol)化合物5、1.22 mL(8.80 mmol)三乙胺、30 mL二氯甲烷依次加入到100 mL单口瓶中, 0 ℃下搅拌均匀, 向体系中慢慢滴加1.23 mL(13.00 mmol)乙酸酐, 滴加完毕后再常温搅拌12 h。反应结束后, 减压蒸馏, 硅胶柱层析, 二氯甲烷/甲醇(V:V=15:1)洗脱, 得1.16 g(5.49 mmol)白色固体6, 收率93%, m.p.201.7~203.0 ℃。
1H NMR(DMSO-d6, 500 MHz) δ:5.02(d, J=3.20 Hz, 1H), 5.00(d, J=3.30 Hz, 1H), 4.34 (s, 1H), 3.64(s, 1H), 3.61~3.56(m, 1H), 3.48~3.42(m, 1H), 2.15(dt, J1=3 Hz, J2=12.7 Hz, 1H), 2.08(dt, J1=3 Hz, J2=12.75 Hz, 1H), 1.80~1.75(m, 2H), 1.75~1.68(m, 2H), 1.44(dt, J1=3 Hz, J2=12.80 Hz, 1H), 1.29(dt, J1=3 Hz, J2=12.75 Hz, 1H); 13C NMR(DMSO-d6, 126 MHz) δ:167.26, 69.46, 68.77, 52.53, 46.72, 31.99, 28.61, 27.75, 23.00, 21.22;IR(KBr, ν/cm-1):3327(O—H), 2930, 2902, 1448(C—H), 1600(C=O); Anal.calcd for C11H17NO3:C 62.54, H 8.11, N 6.63;found:C 62.51, H 8.13, N 6.61。
2.3.5 2-硝基2-氮杂金刚烷-4, 8-二醇二硝酸酯(1)的合成将95%发烟硝酸4.0 mL(84 mmol)和20%发烟硫酸2.5 mL(42 mmol)加入到25 mL茄型瓶中, 0 ℃下搅拌20 min后, 慢慢加入0.6 g(2.84 mmol)化合物6, 再加热到60 ℃反应4 h。反应结束后, 将体系迅速倒入冰水中, 用二氯甲烷萃取(3×15 mL), 合并有机相, 再用饱和碳酸氢钠溶液洗涤, 分液, 水相继续用二氯甲烷萃取, 合并有机相, 用无水硫酸钠干燥, 过滤, 减压蒸馏。硅胶柱层析, 石油醚/乙酸乙酯(V:V=5:1)洗脱, 得0.69 g(2.27 mmol)白色固体1, 收率81%, m.p.107.6~109.1 ℃。
1H NMR(CDCl3, 500 MHz) δ:5.09(t, J=2.85 Hz, 2H), 5.07~5.03(m, 2H), 2.37~2.30 (m, 2H), 2.27(dt, J1=2.85 Hz, J2=14.3 Hz, 2H), 1.98(dt, J1=2.95 Hz, J2=14.1 Hz, 2H), 1.96(t, J=2.94 Hz, 2H); 13C NMR(CDCl3, 126 MHz) δ:53.55, 51.67, 27.98, 27.56, 24.04;IR(KBr, ν/cm-1):2947, 1447(C—H), 1627, 1271(O—NO2), 1532, 1271(N—NO2); Anal.calcd for C9H12N4O8:C 35.53, H 3.98, N 18.42;found:C 35.48, H 3.95, N 18.49。
3 结果与讨论 3.1 双环[3.3.1]壬烷-2, 6-二烯(3)的合成路线关于双环[3.3.1]壬烷-2, 6-二烯的合成, 本研究尝试过采用Bishop等人[24]的三步合成法(见Scheme 2):先用NaBH4将双环[3.3.1]壬烷-2, 6-二酮还原成相应的二醇7, 再将羟基进行磺酸酯化得到化合物8, 再将8溶解在有机强碱DBU中高温热消除得到双环[3.3.1]壬烷-2, 6-二烯。此法第一、二步收率较高, 分别为90%、92%;但是热消除反应收率较低, 仅72%, 总收率为60%。而文献[22]中的两步法后处理简单:第一步腙化反应后处理抽滤、洗涤即可, 第二步消除反应后处理酸洗、水洗、减压蒸馏即可。并且两步法收率更高, 腙化、消除两步总收率达65%。因此本研究选用两步法合成化合物3。

|
Scheme 2 |
本研究以硝硫混酸为硝化剂, 对2-乙酰基-2-氮杂金刚烷-4, 8-二醇的羟基和乙酰基进行硝化, 考察了n(H2SO4):n(HNO3)、反应温度、反应时间对硝化反应的影响。
3.2.1 硫酸与硝酸摩尔比对硝化反应收率的影响控制n(6):n(HNO3)=1:30, 反应温度60 ℃, 反应4 h, 考察了不同的硫酸与硝酸摩尔比对硝化反应收率的影响, 结果见表 1。由表 1可知, 随着硫酸的增加, 反应收率呈先增加后减少的趋势, 在n(H2SO4):n(HNO3)=1:2时, 硝化反应收率最高。分析可能的原因是:当硫酸的量过少时, 硝酸转化为硝酰阳离子的浓度低, 导致硝化能力不强, 硝化不彻底, 收率较低; 当硫酸的量过大时, 体系的酸度增大, 导致氮杂金刚烷骨架的分解[25], 故收率缓慢下降。因此, 硫酸与硝酸的最佳摩尔比是1:2。
| 表 1 硫酸与硝酸摩尔比对硝化反应收率的影响 Tab.1 Effect of the molar ratio between H2SO4 and HNO3 on the yield of nitrolysis reaction |
控制n(H2SO4):n(HNO3)=1:2, 反应4 h, 考察了不同的反应温度对硝化反应收率的影响, 结果见表 2。由表 2可知, 随着反应温度的增加, 反应收率呈先增加后逐渐减少的趋势, 在反应温度为60 ℃时, 硝化反应收率最高, 随后再增加反应温度, 收率反而下降, 这可能是因为, 通常情况, 温度越高硝化体系硝化能力越强, 故刚开始温度越高, 收率增加; 但温度太高, 在强酸性条件下氮杂金刚烷发生开环分解的副反应也会相应增加[25], 导致收率下降。因此, 最佳反应温度为60 ℃。
| 表 2 反应温度对硝化反应收率的影响 Tab.2 Effect of reaction temperature on the yield of nitrolysis reaction |
控制n(H2SO4):n(HNO3)=1:2, 反应温度为60 ℃, 考察了不同的反应时间对硝化反应收率的影响, 结果见表 3。由表 3可知, 随着反应时间的增加, 硝化反应收率增加, 反应4 h收率高达81%, 继续延长反应时间, 收率无明显变化。因此, 最佳反应时间为4 h。
| 表 3 反应时间对硝化反应收率的影响 Tab.3 Effect of reaction time on the yield of nitrolysis reaction |
采用SDT Q600 DSC-TGA同步热分析仪, 氮气流量30 mL·min-1, 温度范围50~300 ℃, 样品量为0.3100 mg, 升温速率10 ℃·min-1条件下得到2-硝基-2-氮杂金刚烷-4, 8-二醇二硝酸酯的TG-DSC谱图如图 1所示。TG曲线表明, 样品从170.0 ℃开始分解, 当温度达到260.0 ℃, 失重为60.5%。该过程中化合物剧烈分解, 由于是负氧平衡, 因此会有单质碳生成。当温度继续升高时, 失重继续缓慢增加, 这可能是单质碳与二氧化碳反应生成了一氧化碳。DSC曲线表明, 该化合物有一个吸热峰和一个放热峰。吸热峰峰型尖锐, 峰值温度为106.5 ℃, 与化合物的熔点(108.4 ℃)基本相吻合, 表明样品经过吸热熔融相变过程; 放热峰峰型较宽, 峰值温度为193.7 ℃。这表明2-硝基-2-氮杂金刚烷-4, 8-二醇二硝酸酯在受热过程中先熔融后分解, 热稳定性良好。

|
图 1 2-硝基-2-氮杂金刚烷-4, 8-二醇二硝酸酯的TG-DSC曲线 Fig.1 TG-DSC curves of 2-nitro-2-azaadamantane-4, 8-diyl dinitrate |
爆速(D)、爆压(p)等是衡量炸药能量特性和爆轰特性的重要指标, 对设计新型含能材料具有重要参考价值, 因此估算爆轰性能参数在理论和实践中均具有重要的意义。Kamlet-Jacobs公式[26]是计算CaHbOcNd类高能量密度化合物爆速、爆压最常用、最简易的方法。该计算公式如下:
| $ \begin{array}{l} D = 1.01{(N{\overline M ^{1/2}}{Q^{1/2}})^{1/2}}\left( {1 + 1.3\rho } \right)\\ p = 1.558{\rho ^2}N{\overline M ^{1/2}}{Q^{1/2}} \end{array} $ |
式中, D为爆速, km·s-1; p为爆压, GPa; N为每克炸药爆轰生成气体的摩尔数, mol·g-1; M — 为气体产物的平均摩尔质量, g·mol-1; Q为每克炸药的爆轰化学能, J·g-1; ρ为理论密度, g·cm-3。
经计算得, 化合物1的密度为1.71 g·cm-3, 爆速为5780 m·s-1, 爆压为11.0 GPa, 性能参数优于1, 3, 5-三硝基金刚烷[18]。
4 结论(1) 以双环[3.3.1]壬烷-2, 6-二酮为原料, 经腙化、消除、环氧化、环合、乙酰化、硝化, 合成了一种新型硝基笼形化合物——2-硝基-2-氮杂金刚烷-4, 8-二醇二硝酸酯, 总收率27%, 并利用IR、NMR、元素分析等对其进行了表征。
(2) 以硝硫混酸为硝化剂, 优化了硝化反应的工艺条件, 确定的适宜的反应条件:n(H2SO4):n(HNO3)= 1:2, 反应温度为60 ℃, 反应时间为4 h, 硝化反应收率为81%。
(3) 利用TG和DSC研究了2-硝基-2-氮杂金刚烷-4, 8-二醇二硝酸酯的热行为, 发现其从170.0 ℃开始分解, 分解峰值为193.7 ℃, 表明热稳定性较好。
(4) 预估了2-硝基-2-氮杂金刚烷-4, 8-二醇二硝酸酯的物化和爆轰性能, 其密度为1.71 g·cm-3, 爆速为5780 m·s-1, 爆压为11.0 GPa, 可以用于炸药及推进剂的配方中。
| [1] |
刘卅, 郭建维. 金刚烷的结构、溶解性及热力学性质[J].
含能材料, 2006, 14(6): 485-490. LIU Sa, GUO Jian-wei. Structure analysis, solubility and thermodynamics properties of adamantane[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2006, 14(6): 485-490. |
| [2] |
许晓娟, 肖鹤鸣, 居学海, 等. 多硝基金刚烷红外光谱和热力学性质的理论研究[J].
含能材料, 2005, 13(1): 40-44. XU Xiao-juan, XIAO He-min, JU Xue-hai, et al. Theoretical study on the vibrational spectra, thermodynamic properties for polynitroadamantanes[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2005, 13(1): 40-44. |
| [3] |
王飞, 许晓娟, 肖鹤鸣, 等. 多硝基金刚烷生成热和稳定性的理论研究[J].
化学学报, 2003, 61(12): 1939-1943. WANG Fei, XU Xiao-juan, XIAO He-ming, et al. Theoretical studies on heat of formation and stability for polynitroadamantanes[J]. Acta Chimica Sinica, 2003, 61(12): 1939-1943. DOI:10.3321/j.issn:0567-7351.2003.12.010 |
| [4] |
Skare D, Suceska M. Polynitroadamantanes-new caged high-energetic molecules[J].
Kemija u industriji, 1995, 44(12): 511-518. |
| [5] |
杜耀, 王艳红, 李雅津, 等. 多硝基金刚烷的合成及其理论研究进展[J].
化学推进剂与高分子材料, 2014, 12(2): 57-63. DU Yao, WANG Yan-hong, LI Ya-jin, et al. Progress in synthesis and theory research of polynitroadamantanes[J]. Chemical Propellants & Polymeric Materials, 2014, 12(2): 57-63. |
| [6] |
Sollott G P, Gilbert E E. A facile route to 1, 3, 5, 7-tetraaminoadamantane:synthesis of 1, 3, 5, 7-tetranitroadamantane[J].
J Org Chem, 1980, 45: 5405-5408. DOI:10.1021/jo01314a051 |
| [7] |
罗军, 李娅琼. 用改进的Sollott法合成1, 3, 5, 7-四硝基金刚烷[J].
火炸药学报, 2011, 34(5): 13-15. LUO Jun, LI Ya-qiong. Synthesis of 1, 3, 5, 7-tetranitroadamantane via modified sollott's route[J]. Chinese Journal of Explosives and Propellants, 2011, 34(5): 13-15. |
| [8] |
Gilbert E E. 1, 3, 5, 7-Tetranitroxyadamantane: US, 4476060[P]. 1983.
|
| [9] |
Archibald T G, Baum K. Synthesis of polynitroadamantanes:oxidations of oximinoadamantanes[J].
J Org Chem, 1988, 53(20): 4646-4649. |
| [10] |
凌亦飞, 孙露, 罗军. 2, 2, 6, 6-四硝基金刚烷的合成与表征[J].
含能材料, 2015, 23(9): 877-881. LING Yi-fei, SUN Lu, LUO Jun. Synthesis and characterization of 2, 2, 6, 6-tetranitroadamantane[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2015, 23(9): 877-881. DOI:10.11943/j.issn.1006-9941.2015.09.008 |
| [11] |
Dave P R, Ferraro M. Synthesis of 2, 2, 4, 4-tetranitroadamantane[J].
J Org Chem, 1990, 55: 4459-4461. DOI:10.1021/jo00301a047 |
| [12] |
孙露, 凌亦飞, 张萍萍, 等. 2, 2, 4, 4-四硝基金刚烷的合成与表征[J].
含能材料, 2014, 22(4): 447-453. SUN Lu, LING Yi-fei, ZHANG Ping-ping, et al. Synthesis and characterization of 2, 2, 4, 4-tetranitroadamantane[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2014, 22(4): 447-453. |
| [13] |
Paritosh R D. The synthesis and complete 1H and 13C NMR spectral assignment of 2, 2, 4, 4, 6, 6-hexanitroadamantane and its precursor nitroketones by 2D NMR spectroscopy[J].
Tetrahedron, 1992, 48: 5839-5846. DOI:10.1016/S0040-4020(01)90175-9 |
| [14] |
张萍萍, 凌亦飞, 孙露, 等. 2, 2, 4, 4, 6, 6-六硝基金刚烷的合成、表征及晶体结构[J].
含能材料, 2014, 22(5): 646-653. ZHANG Ping-ping, LING Yi-fei, SUN Lu, et al. Synthesis, characterization and crystal structure of 2, 2, 4, 4, 6, 6-hexanitroadamantane[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2014, 22(5): 646-653. |
| [15] |
Theodore A, Lida Q. Synthesis of 1, 2, 2-trinitroadamantane[J].
J Org Chem, 1995, 60: 1895-1896. DOI:10.1021/jo00111a062 |
| [16] |
LING Yi-fei, REN Xiao-li, LAI Wei-peng, et al. 4, 4, 8, 8-tetranitroadamantane-2, 6-diyl dinitrate:a high-density energetic material[J].
European Jounal of Organic Chemistry, 2015(7): 1541-1547. |
| [17] |
冯增国. 多面体含能材料的爆速和爆压预估[J].
含能材料, 1993, 1(2): 42-48. FENG Zeng-guo. Predicting detonation velocity and pressure of polyhedron energetic materials[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 1993, 1(2): 42-48. |
| [18] |
魏真, 李加荣, 张奇, 等. 金刚烷类高能量密度燃料研究进展[J].
含能材料, 2014, 22(2): 170-176. WEI Zhen, LI Jia-rong, ZHANG Qi, et al. Review on diamondoids as hifh energetic density fuels[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2014, 22(2): 170-176. |
| [19] |
Severin T, Baetz D, Kraemer H. Synthesis of trinitroazaadamantane derivatives[J].
Chemische Berichte, 1971, 104(3): 950-953. DOI:10.1002/(ISSN)1099-0682 |
| [20] |
Nielsen A T, Chafin A P, Christian S L, et al. Synthesis of polyazapolycyclic caged polynitramines[J].
Tetrahedron, 1998, 54(39): 11793-11812. DOI:10.1016/S0040-4020(98)83040-8 |
| [21] |
LING Yi-fei, ZHANG Ping-ping, SUN Lu, et al. Efficient synthesis of 2, 2, 4, 4, 6, 6-hexanitroadamantane under mild conditions[J].
Synthesis, 2014, 46: 2225-2233. DOI:10.1055/s-00000084 |
| [22] |
Mayr M, Bataille C J, Gosiewska S, et al. Synthesis and rhodium complexation of enantiomerically enriched bicyclo[3.3.1]nona-2, 6-diene[J].
Tetrahedron:Asymmetry, 2008, 19: 1328-1332. DOI:10.1016/j.tetasy.2008.05.009 |
| [23] |
Henkel J G, Faith W C, Hane J T. A general synthesis of N-substituted 2-azaadamantanes and their 4, 8-disubstituted derivatives[J].
J Org Chem, 1981, 46(17): 3483-3486. DOI:10.1021/jo00330a020 |
| [24] |
Bishop R, Choudhury S, Dance I. The synthesis and crystal structures of exo-2, exo-6-dihydroxybicyclo[3.3.l]nonane and exo-2, exo-6-dihydroxy-2, 6-diethylbicyclo[3.3.1]nonane which do not contain the host helical canals of crystalline exo-2, exo-6-dihydroxy-2, 6-dimethylbicyclo[3.3.1]nonane[J].
Journal of the Chemical Society, Perkin Transactions 2, 1982, 9: 1159-1168. |
| [25] |
史楠, 胡炳成, 吕春绪. 小分子法合成1, 5-二乙酰基-3, 7-二硝基-1, 3, 5, 7-四氮杂辛烷工艺研究[J].
火炸药学报, 2013, 36(2): 13-19. SHI Nan, HU Bing-cheng, LüChun-xu. Study on the process of synthesizing 1, 5-diacetyl-3, 7-dinitro-1, 3, 5, 7-tetraazacyclooctane by small-molecule method[J]. Chinese Journal of Explosives and Propellants, 2013, 36(2): 13-19. |
| [26] |
Kamlet M J, Jacobs M J. Chemistry of detonation I a simple method for calculation detonation properties of C—H—N—O explosives[J].
Journal of Chemical Physics, 1968, 48(1): 23-35. DOI:10.1063/1.1667908 |
2-Nitro-2-azaadamantane-4, 8-diyl dinitrate was synthesized from bicyclo[3.3.1]nonane-2, 6-dione. The optimal nitration reaction conditions was obtained and the thermal stability of target product was studied by thermogravimetry (TG) and differential scanning calorimetry(DSC).The detonation properties were predicted by Kamlet-Jacobs formula.