




2. 盐城工学院化学与化工学院, 江苏 盐城 224051
2. School of Chemical and Biological Engineering, Yancheng Institute of Technology, Yancheng 224051, China
随着现代武器系统的不断发展, 高能钝感炸药成为炸药研发领域的热点。目前, 许多国家都致力于研制高能钝感炸药, 以降低弹药因外界刺激自发爆炸的几率。2, 6-二氨基-3, 5-二硝基吡啶-1-氧化物(ANPyO)爆炸性能、机械感度及热安定性与三氨基三硝基苯(TATB)相近[1-2], 但制造成本显著低于TATB, 故ANPyO作为潜在高能钝感炸药具有广阔的应用前景, 有关ANPyO合成路线及性能的研究受到国内外研究人员的广泛关注[3-10]。
晶体形貌是影响炸药机械感度的一个重要参数, 其主要受晶体内部结构和外部环境的影响, 重结晶法的外部环境主要包括溶剂、溶液过饱和度、溶液pH值、温度、粘度及结晶速度等, 其中溶剂所起的作用至关重要。通过改变重结晶溶剂种类、溶剂浓度、温度及搅拌速率等工艺条件可以控制炸药晶体形貌、减少晶体缺陷、提高晶体品质、降低炸药感度、提高炸药安全性能, 但工艺优化过程还主要依赖实验筛选, 耗时费力。
计算机技术的发展及晶体生长理论知识体系的不断完善, 使得从分子尺度模拟晶体生长过程得以实现, 随之出现了一系列的晶体形貌预测模型, 如考虑晶面间距对生长速率影响的Bravis-Friedel-Donnay-Harker(BFDH)法则[11], 依据晶体平衡形态由0K时表面最小表面能决定的Equilibrium Morphology法[12], 晶面螺旋生长的Burton-Cabrera-Frank(BCF)机制[13]及基于晶体晶面生长速率与附着能绝对值成正比的附着能(AE)模型[14]等, 国内外学者基于晶体形貌预测模型成功地预测了医药品[15]、炸药[16]、有机颜料[17]及肾结石[18]晶体形貌。采用分子动力学(MD)模拟预测真空晶体形貌及溶剂对晶体形貌的影响不仅可以给出更多溶剂和晶体相互作用的微观信息, 而且可以减少实验筛选规模、节约时间和财力, 加快新产品的研发。
目前, 对ANPyO的研究多侧重合成路线改进、晶型控制的实验研究, 而有关ANPyO结晶理论研究少。刘祖亮[7]等采用密度泛函理论计算了ANPyO晶体的相关性质及理论晶格能, 结果表明ANPyO晶体导带非常平坦, 而价带凸凹不平; 分子中的C—NO2键和N—O键比较弱, 在外界刺激下容易断裂, 且较之TATB, ANPyO对机械力更敏感, 计算结果与实验结果吻合。居学海[8]等采用第一性原理方法研究了ANPy及ANPyO双聚体的结构及结合能, 计算结果表明ANPy及ANPyO双聚体中皆存在氢键, 且ANPy双聚体中的氢键强度比ANPyO双聚体中的大, 这与氢键距离一致。但是, 两者的研究中均未涉及ANPyO晶体形貌的预测及溶剂对ANPyO晶体形貌的影响。基于此, 本研究拟借助Material Studio(MS)软件包[19], 采用AE模型预测ANPyO的真空晶体形貌, 确定其形态学上重要的生长晶面, 并建立ANPyO主要生长晶面和溶剂N, N-二甲基甲酰胺(DMF)的双层结构模型, 在正则系综(NVT)下对双层模型进行MD模拟, 获得ANPyO主要生长晶面与溶剂分子的相互作用能, 进而运用修正AE模型预测ANPyO在溶剂中的晶体形貌, 并与实验观察结果进行对比。此外, 通过分析DMF和ANPyO的径向分布函数, 探讨溶剂和ANPyO相互作用本质, 为ANPyO结晶溶剂的选择提供一定的理论基础。
2 理论及模拟方法 2.1 AE模型及修正的AE模型AE模型是Hartman和Bennema基于周期键链(PBC)理论提出来的[20], 该模型定义厚度为dhkl的晶片附着在一块正在生长的晶体表面(h k l)时所释放的能量为附着能Eatt, 晶片附着所需时间随附着能绝对值的增大而减小, 故而晶面法向生长速度Rhkl与晶面附着能绝对值成正比, 具有最低附着能绝对值的晶面生长速度最慢, 具有最大形态学重要性。根据各晶面附着能, 计算获得晶体生长速率, 确定每个晶面的面心距离, 再借由Wulff图分析这些信息, 就可以预测晶体的真空晶体形貌。
| $ {R_{{\rm{hkl}}}}\infty \left| {{E_{{\rm{att}}}}} \right| $ | (1) |
溶液中生长的晶体不同晶面与溶剂分子的相互作用能Eint亦不同, 使得溶液中各晶面的附着能与其真空中的值不同, 晶面法向生长速度发生改变, 晶体形貌也随之发生一定的变化。为获得溶剂与ANPyO晶体晶面的Eint, 本模拟计算采用双层结构模型, 即为上下两层构成的3D周期结构, 其上层是溶剂分子、下层是具有一定厚度的ANPyO晶面。Eint可按照式(2)计算获得:
| $ {E_{{\rm{int}}}} = {E_{{\rm{tot}}}}-{E_{{\rm{surf}}}}-{E_{{\rm{solv}}}} $ | (2) |
式中, Etot为双层结构模型的平均单点能; Esurf为双层结构模型中晶面的平均单点能; Esolv为双层结构模型中溶剂的平均单点能。
鉴于溶剂与晶面存在相互作用, 对晶面的附着能有一定的影响, 为考察溶剂对ANPyO晶体形貌的影响, 本研究定义修正的附着能E′att为:
| $ {E^\prime _{{\rm{att}}}} = {E_{{\rm{att}}}}-{E_{\rm{s}}} $ | (3) |
式中, Es为附着能校正项, 涉及溶剂与晶面的相互作用能Eint、晶体单元胞晶面溶剂可达面积Aacc及双层结构模型总晶面面积Abox, 如式(4)所示:
| $ {E_{\rm{s}}} = {E_{{\rm{int}}}} \times {A_{{\rm{acc}}}}/{A_{{\rm{box}}}} $ | (4) |
根据式(1), 溶液中晶面法向生长速度R′hkl与E′att绝对值成正比:
| $ {R^\prime_{{\rm{hkl}}}} \propto \left| {{E^\prime_{{\rm{att}}}} } \right| $ | (5) |
溶液中晶面的E′att绝对值越小, 则此晶面生长越慢, 其在晶体形貌中呈现最大形态学重要性。
2.2 模型构建借助MS(Accelrys Inc., USA)软件包, 根据Hollins等[21]的实验数据构建ANPyO晶胞的初始构型。ANPyO晶体属于单斜晶系、C2/c空间群, 每个晶胞内有2个对称独立的分子, 结晶密度为1.878 g·cm-3, 晶格参数:a=1.4864 nm, b=0.7336 nm, c=0.7509 nm, β=111.67°。采用Discover模块中的Smart Minimizer方法、Compass力场对ANPyO晶胞进行能量最小化(EM), 优化晶胞参数。在此基础上, 采用Morphology模块中的AE模型预测ANPyO晶体的真空晶体形貌, 获得ANPyO主要生长晶面。
对优化过的ANPyO晶胞进行“切割分面”, 获得其主要生长晶面(1 1 0), (1 0 0), (1 0 -1)及(1 1 -2)面, 并分别扩展成2.253 nm×2.486 nm×3.633 nm, 2.201 nm×2.253 nm×3.460 nm, 2.934 nm×2.792 nm×2.915 nm及2.768 nm×3.315 nm×3.189 nm的3D周期结构, 其中(1 1 0)和(1 0 0)超晶胞含54个ANPyO分子(1080个原子)、(1 0 -1)和(1 1 -2)超晶胞含64个ANPyO分子(1280个原子)。据已有研究结果[5], N, N-二甲基甲酰胺(DMF)可作为ANPyO的重结晶溶剂, 在Discover模块构建DMF分子, 采用Smart Minimizer方法、Compass力场对DMF分子进行MM; 采用Amorphous Cell模块构建与(1 1 0), (1 0 0), (1 0 -1)及(1 1 -2)面的3D周期结构尺寸相符的DMF溶剂层(100个DMF分子, 密度为0.9487 g·cm-3)。对溶剂层进行EM后, 在288 K进行MD模拟, 模拟时间100 ps, 步长1 fs, 温度控制采用Andersen方法[22]。使用分层建模工具构建DMF/ANPyO双层结构初始模型, 其中上层为经MD模拟的DMF溶剂、下层分别为已构建好的ANPyO(1 1 0), (1 0 0), (1 0 -1)及(1 1 -2)面的3D周期结构, 周期箱在c方向添加2 nm的真空层, 如图 1所示。

|
图 1 DMF/ANPyO晶面双层模型初始结构侧视图 Fig.1 Equilibrium configurations of DMF/ANPyO crystal faces |
双层结构初始模型通过EM后, 在Compass力场下, 选取NVT系综, 温度设为288 K, 温度控制采用Anderson[22]方法, 用Velocity Verlet积分法求解牛顿运动方程, 原子初始速度按Boltzmann随机分布方法确定。模拟过程中Coulomb和van der Waals(vdW)长程非键作用力分别采用Ewald和Atom based方法计算求得, 其中vdW力采用球形截断法进行长程校正、截断半径取为0.95 nm, 截断距离之外的分子间相互作用按平均密度近似方法进行校正。模拟时间步长为1 fs, 模拟时间为500 ps, 其中前200 ps用于体系的热力学平衡、后300 ps用于体系的统计分析, 每500 fs输出一帧轨迹。
3 结果及讨论 3.1 模拟力场的选择Compass力场是一个由凝聚态性质、孤立分子的各种从头算和经验数据等参数化并验证的从头算力场, 广泛适用于大多数常见无机物、有机物及聚合物分子, 能在大范围内准确预测孤立体系和凝聚态体系的构型和热力学性质。力场优劣是影响计算结果可靠性的重要因素, 因而在研究具体体系时, 力场适用性的检验是非常重要的环节。本研究在模拟计算之前, 分别采用Compass力场、Pcff力场对ANPyO晶胞进行优化, 表 1列出ANPyO晶胞参数的计算值及实验值。
| 表 1 ANPyO晶胞参数理论计算值与实验值 Tab.1 Calculated and experimental values of ANPyO cell parameters |
分析表 1可知, 由Compass力场和Pcff力场计算的参数α,γ值皆为90°, 与实验值完全吻合, 两者计算的参数β误差分别为0.29%、-1.08%。此外, Compass力场计算除了参数c的误差稍大(-7.87%), 参数a, b的误差都较小, 而Pcff力场计算的参数a, b, c的误差都较大, 故综合考虑, 本计算选择Compass力场。
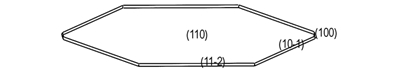
3.2 真空中ANPyO的晶体形貌图 2给出了采用AE模型(电荷分布及力场分别采用Forcefield方法及Compass力场)预测的真空中ANPyO的晶体形貌, 表 2列出了ANPyO主要生长晶面的相关参数。

|
图 2 AE模型预测真空中ANPyO的晶体形貌 Fig.2 ANPyO crystal morphology in vacuum predicted by the AE model |
| 表 2 ANPyO主要生长晶面的参数1) Tab.2 The parameters of the main crystal faces of ANPyO |
如图 2所示, 由AE模型预测的真空中ANPyO的晶体形貌接近椭圆, 长径比为2.349, 其主要生长晶面为(1 1 0)面, (1 0 0)面, (1 0 -1)面及和(1 1 -2)面。
分析表 2可知, ANPyO在真空中的主要生长晶面为(1 1 0)面, (1 0 0)面, (1 0 -1)面及(1 1 -2)面, 其中多重度为2的(1 1 0)面的表面积占总面积40.70%, 所占比例最大且(1 1 0)面具有最大的晶面间距(d110=0.691 nm); 多重度为4的(1 0 -1)面的表面积占总面积35.69%;(1 1 0)面与(1 0 -1)面是是影响ANPyO晶体形态最重要的晶面, 其次为(1 0 0)面和(1 1 -2)面, 表面积分别占总面积的17.38%和6.24%。
3.3 溶剂N, N-DMF与ANPyO晶面的相互作用能及其本质溶剂DMF与ANPyO的4个主要生长晶面分别构成4个双层结构模型, 经能量最小化后进行MD模拟, 200 ps后体系达到热力学平衡。图 3给出了DMF/ANPyO晶面的平衡构型侧视图。

|
图 3 溶剂DMF/ANPyO晶面的平衡构型侧视图 Fig.3 The side view of the starting conformations for DMF/ANPyO crystal faces interfacial model |
分析图 3可知, 溶剂DMF/ANPyO晶面的双层模型经MD模拟后, 溶剂DMF分子扩散到ANPyO的(1 1 0)面, (1 0 0)面, (1 0 -1)面及(1 1 -2)面并吸附在晶面上, 说明溶剂DMF与ANPyO的这4个晶面间存在较强的相互作用。表 3列出了溶剂DMF/ANPyO晶面双层结构模型的相互作用能及其组成。
| 表 3 溶剂DMF/ANPyO晶面双层结构模型的相互作用能1) Tab.3 The interaction energies of the interfacial model of DMF/ANPyO face |
依据式(2), 溶剂DMF与ANPyO(1 1 0)面, (1 0 0)面, (1 0 -1)面及(1 1 -2)面的相互作用能分别为-504.76, -334.89, -593.58 kJ·mol-1和-1045.46 kJ·mol-1, 皆为负值, 说明溶剂DMF在晶面的吸附是一个放热过程, 在热力学上属于自发过程。在溶剂DMF环境下, ANPyO粒子要沉积在晶面上必须首先排除晶面上的DMF分子, 此过程需要消耗能量, 导致晶面生长速度降低。溶剂DMF与晶面相互作用能的绝对值越大, 则DMF在ANPyO晶面的吸附能力越强。DMF的存在会影响ANPyO晶面的生长速度, 进而影响其晶体形貌。
分析表 3可知, DMF与ANPyO晶面的相互作用由强到弱的顺序为:(1 1 -2)>(1 0 -1)>(1 1 0)>(1 0 0), DMF与ANPyO晶面的相互作用能近似等于非键能, 即范德华力和静电力之和, 其中范德华力所占比例较大, 但两者均很重要。在Compass力场中, 氢键包含在静电力中, 没有独立显示项。
通过对整个平衡后的轨迹进行径向分布函数g(r)-r分析, 可进一步揭示溶剂DMF与ANPyO晶面相互作用的方式及本质。g(r)-r可理解为给定某个粒子的坐标, 其它粒子出现在距离该粒子为r的、无限薄的球壳层的几率。g(r)可用于固体、液体的结构及特殊相互作用的研究, 如氢键[23]。在g(r)-r图中, 通常r=0.11~0.31 nm为氢键、r=0.31~0.50 nm为vdW作用, 大于0.50 nm的vdW作用比较微弱。图 4绘制了ANPyO(1 1 -2)面的周期箱中最上层ANPyO分子中的H原子与溶剂DMF分子中的N原子、O原子的g(r)N—H-r图、g(r)O—H-r图。分析DMF与ANPyO(1 1 0)面、(1 0 0)面及(1 0 -1)面平衡后的轨迹, 得到与ANPyO(1 1 -2)面类似的g(r)-r图。

|
图 4 DMF与ANPyO(1 1 -2)面的径向分布函数 Fig.4 The radial distribution function of DMF and ANPyO(1 1 -2) face |
分析g(r)O—H-r图, 在r=0.21~0.22 nm范围内出现最高峰值, 说明DMF分子中的O原子可以与ANPyO(1 1 -2)面的H原子形成氢键。在g(r)N—H-r图中, r=0.43~0.44 nm范围内出现最高峰值, 说明DMF分子中N原子与ANPyO(1 1 -2)面的H原子间存在较强的vdW作用。溶剂DMF与ANPyO晶面的相互作用主要源自DMF分子中的O、N原子分别与ANPyO晶面的H原子形成的氢键和vdW作用, 和溶剂与晶面相互作用能计算获得的结果一致。
3.4 溶剂DMF对ANPyO晶体形貌的影响晶体平衡形态理论认为, 在结晶过程中, 结构基元结合到晶体表面上时所释放的结合能为附着能, 结合所需时间随释放的结合能增大而减小, 故晶面的法向生长速度将随晶面附着能绝对值的增大而增大。为考察溶剂DMF对ANPyO晶体形貌的影响, 本研究引入修正附着能E′att, 其计算涉及溶剂与晶面的相互作用能Eint、晶体单元胞晶面溶剂可达面积Aacc及双层结构模型总晶面面积Abox, 具体计算参照式(3)和式(4), 计算结果列于表 4。
| 表 4 ANPyO晶面的修正附着能及法向生长速度 Tab.4 The modified attachment energies and the normal growth rate of ANPyO crystal faces |
具有最低附着能绝对值的表面生长最慢, 具有形态学上的重要性。炸药晶体生长过程中, 各个晶面生长速率不同, 生长快的晶面终将消失, 最后显露的晶面决定了晶体的形貌。分析表 4可知, 溶剂DMF对ANPyO晶体的不同晶面的附着能大小的影响不同, 在考虑能量修正项后, ANPyO晶面的修正附着能绝对值顺序为(1 1 0)<(1 1 -2)<(1 0 -1)<(1 0 0)。(1 1 0)面具有最低附着能绝对值, 法向生长速度最慢, 具有形态学上的重要性, 是ANPyO的主要生长晶面; 而(1 0 0)面、(1 0 -1)面及(1 1 -2)面的法向生长速度较快, 故这3个晶面的面积显著减小, 甚至(1 0 0)面有消失的趋势。依据ANPyO在溶剂DMF中的修正附着能, 借助Morphology模块预测的ANPyO在溶剂DMF中的晶体形貌见图 5。

|
图 5 修正的AE模型预测ANPyO在溶剂DMF中的晶体形貌 Fig.5 Crystal morphology of ANPyO in DMF predicted by the modified AE model |
如图 5所示, 修正的AE模型预测的ANPyO在溶剂DMF中的晶体形貌接近片状, 与刘祖亮等[5]实验所得结果一致。其长径比为78.72, (1 1 0)面、(1 0 -1)面、(1 1 -2)面及(1 0 0)面的表面积分别占总面积的96.44、2.15、1.28及0.13。
3.5 溶剂DMF分子在ANPyO晶面的扩散系数对晶体形貌的影响溶剂在晶面的扩散能力与溶剂分子在晶面的扩散系数(D)有关[24], 根据爱因斯坦扩散方程, 分子的扩散系数可以通过均方根位移(MSD)对时间(t)的微分函数获得, 如式(6)所示:
| $ D = \frac{1}{6}\mathop {\lim }\limits_{{\rm{t}} \to \infty } \frac{d}{{d{\rm{t}}}}\sum\limits_{i = 1}^N {\left\langle {{{\left| {r\left( t \right)-{r_i}\left( 0 \right)} \right|}^2}} \right\rangle } $ | (6) |
式中, ri指粒子的位置矢量。MSD对时间的函数的斜率常用来计算一个粒子在三维空间进行随机布朗运动的扩散系数。MSD的计算可以在Discover模块中通过模拟轨迹的统计分析获得, 图 6给出了288 K下, DMF分子在ANPyO(1 1 0)、(1 0 -1)、(1 1 -2)及(1 0 0)面的均方根位移。

|
图 6 DMF分子在ANPyO(1 1 0)、(1 0 -1)、(1 1 -2)及(1 0 0)面的均方根位移(288 K) Fig.6 The mean square displacement of DMF molecules on the(1 1 0), (1 0 -1), (1 1 -2) and(100) faces at 288 K |
通过图 6中MSD与t函数的斜率可估算出DMF在ANPyO(1 1 0)、(1 0 -1)、(1 1 -2)及(1 0 0)面的扩散系数分别为1.29×10-9, 1.14×10-9, 1.19×10-9及1.07×10-9m2·s-1。结果表明, DMF分子在ANPyO(1 1 0)面的扩散能力最强, 在ANPyO(1 0 0)面的扩散能力最差。
晶面法向生长速度与修正附着能的绝对值成正比, 修正附着能的绝对值越大, 则该晶面的法向生长速度越快, 甚至在生长过程中该晶面会消失; 反之, 晶面的法向生长速度越慢, 成为晶体的主要生长晶面, 在晶体形貌中呈现最大形态学重要性。本研究通过研究溶剂分子在ANPyO晶面的扩散系数与晶面修正附着能的绝对值的关系, 探讨了溶剂DMF在晶面的扩散系数对ANPyO晶体形貌的影响。
分析图 7可知, 溶剂DMF分子在ANPyO晶面的扩散系数与修正附着能绝对值成线性关系。DMF分子在ANPyO(1 1 0)面的扩散系数最大, 即较之其它三个晶面, 溶剂分子更易于迁移至(1 1 0)面, 促使(1 1 0)面的修正附着能的绝对值最小, (1 1 0)面具有最大的表面积, 是ANPyO晶体的主要生长晶面。由此可见, 溶剂DMF在晶面的扩散能力亦影响ANPyO的晶体形貌。

|
图 7 DMF分子在ANPyO晶面扩散系数与修正附着能绝对值的关系(288 K) Fig.7 Relationship between the diffusion coefficient of DMF molecules on ANPyO crystal faces and the absolute value of the corrected attachment energies at 288 K |
通过DMF溶剂层/ANPyO晶面双层结构模型的分子动力学模拟, 借助修正附着能模型研究溶剂对ANPyO晶体形貌的影响, 主要结论如下:
(1) 附着能模型预测ANPyO在真空中的晶体形状接近椭圆, 主要生长晶面为(1 1 0)、(1 0 0)、(1 1 -2)及(1 0 -1)面。考虑能量修正项, ANPyO主要生长晶面的修正附着能绝对值顺序为(1 1 0)<(1 1 -2)<(1 0 -1)<(1 0 0), 在溶剂DMF中, ANPyO晶体形状接近于片状。
(2) 径向分布函数分析表明溶剂DMF和ANPyO晶面的相互作用能主要包括范德华作用, 库仑作用和氢键。
(3) DMF分子在ANPyO晶面的扩散系数计算表明DMF分子在ANPyO(1 1 0)面的扩散能力最强, 在ANPyO(1 0 0)面的扩散能力最差, ANPyO晶体生长形貌亦受DMF分子扩散能力的影响。拟结果为ANPyO结晶溶剂的选择提供一定的理论基础。
| [1] |
Licht H H. Performance and sensitivity of explosives[J].
Propellants, Explosives, Pyrotechnics, 2002, 25(3): 126-132. |
| [2] |
马丛明, 刘祖亮, 许晓娟, 等. 吡啶类含能化合物的合成研究进展[J].
有机化学, 2014, 34: 1288-1299. MA Cong-ming, LIU Zu-liang, XU Xiao-juan, et al. Research progress on the synthesis of energetic pyridines[J]. Chinese Journal of Organic Chemistry, 2014, 34: 1288-1299. |
| [3] |
Badgujar D M, Talawar M B, Asthana S N, et al. Advances in science and technology of modern energetic materials:an overview[J].
Journal of Hazardous Materials, 2008, 151(2): 289-305. |
| [4] |
Ritter H, Licht H H. Synthesis and reaction of dinitrate amino and diaminopyridine[J].
Journal of Heterocycls Chemistry, 1995, 32: 585-590. DOI:10.1002/jhet.v32:2 |
| [5] |
成健, 姚其正, 刘祖亮. 2, 6-二氨基-3, 5-二硝基吡啶-1-氧化物的合成新方法[J].
含能材料, 2009, 17(2): 166-168. CHENG Jian, YAO Qi-zheng, LIU Zu-liang. Synthesis of 2, 6-diamino-3, 5-dinitropyridine-1-oxide[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2009, 17(2): 166-168. |
| [6] |
王艳红, 宋元军, 胡桢, 等. 一个简易的2, 6-二氨基-3, 5-二硝基吡啶-1-氧化物的合成方法[J].
有机化学, 2009, 29(5): 780-783. WANG Yan-hong, SONG Yuan-jun, HU Zhen, et al. A simple method for preparing 2, 6-diamino-3, 5-dinitropyridine-1-oxide[J]. Chinese Journal of Organic Chemistry, 2009, 29(5): 780-783. |
| [7] |
HE Zhi-wei, ZHOU Su-qiu, JU Xue-hai, et al. Computational investigation on 2, 6-diamino-3, 5-dinitropyridine-1-oxide crystal[J].
Journal of Structural Chemistry, 2010(21): 651-656. |
| [8] |
XIE L F, YE C C, JU X H, et al. Theoretical study on dimers of 2, 6-diamino-3, 5-dinitropyridine and its N-oxide[J].
Journal of Structural Chemistry, 2012(4): 659-664. |
| [9] |
Anderson K L, Merwin L H, Wilson W S, et al. 15N chemical shifts in energetic materials:CP/MAS and ab initio studies of aminonitropyridines, aminonitropyrimidines, and their N-oxides[J].
International Journal of Molecular Science, 2002(3): 858-872. |
| [10] |
刘华宁, 郑宇, 邱从礼, 等. 新型炸药2, 6-二氨基-3, 5-二硝基吡啶-1-氧化物的射流冲击感度实验研究[J].
含能材料, 2014, 22(3): 337-342. LIU Hua-ning, ZHENG Yu, QIU Cong-li, et al. Experimental study on jet impact sensitivity of a new explosive 2, 6-diamino-3, 5-dinitropyridine-1-oxide[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2014, 22(3): 337-342. |
| [11] |
Donnay J D H, Harker D. A new law of crystal morphology extending the law of Bravais[J].
Am Mineral, 1937, 22(5): 446-467. |
| [12] |
Titiloye J O, Parker S C, Mann S. Atomistic simulation of calcite surfaces and the influence of growth additives on their morphology[J].
Journal of Crystal Growth, 1993, 131(3): 533-545. |
| [13] |
Myers-Beaghton A K, Vvedensky D D. Generalized Burton-Cabrera-Frank theory for growth and equilibr-ation on stepped surfaces[J].
Physical Review A, 1991, 44(4): 2457 DOI:10.1103/PhysRevA.44.2457 |
| [14] |
Clydesdale G, Hammond R B, Ramachandran V, et al. Molecular modelling of the morphology of organic crystals in the presence of impurity species:Recent applications to naphthalene, phenanthrene, and caprolactam crystals[J].
Molecular Crystals and Liquid Crystals, 2005, 440(1): 235-257. DOI:10.1080/15421400590958566 |
| [15] |
Coombes D S, Catlow C R A, Gale J D, et al. Theoretical and experimental investigations on the morphology of pharmaceutical crystals[J].
J Pharm Sci, 2002, 91: 1652-1658. DOI:10.1002/jps.10148 |
| [16] |
Ter Horst J H, Kramer H J M, van Rosmalen G M, et al. Molecular modelling of the cryst -allization of polymorphs.Part Ⅰ:The morphology of HMX polymorphs[J].
J Cryst Growth, 2002, 237: 2215-2220. |
| [17] |
Erk P. Crystal design of organic pigments-a prototype discipline of materials science[J].
Curr Opin Solid State Mater Sci, 2001(5): 155-160. |
| [18] |
Millan A. Crystal growth shape of whewellite polymorphs:influence of structure distortions on crystal shape[J].
Crys Growth Des, 2001(1): 245-254. |
| [19] |
Accelrys Software Inc. Materials Studio 3. 0[CP]. Discover/Accelrys Software Inc, San Diego, California, 2004.
|
| [20] |
Hartman P, Bennema P. The attachment energy as a habit controlling factor[J].
Journal of Crystal Growth, 1980, 49: 145-156. DOI:10.1016/0022-0248(80)90075-5 |
| [21] |
Hollins R A, Merwin L H, Nissan R A, et al. Aminonitropyridines and their N-oxides[J].
Journal of Heterocycls Chemistry, 1996, 33: 895-904. DOI:10.1002/jhet.v33:3 |
| [22] |
Andersen H C. Molecular dynamics simulations at constant pressure and/or temperature[J].
The Journal of Chemistry Physical, 1980, 72: 2384-2393. DOI:10.1063/1.439486 |
| [23] |
孙婷, 刘强, 肖继军, 等. CL-20/HMX共晶及其为基PBX界面作用和力学性能的MD模拟研究[J].
化学学报, 2014, 72: 1036-1042. SUN Ting, LIU Qiang, XIAO Ji-jun, et al. Molecular dynamics simulation of interface interactions and mechanical properties of CL-20/HMX cocrystal and its based PBXs[J]. Acta Chim Sinica, 2014, 72: 1036-1042. |
| [24] |
刘蓓, 廉源会, 李智, 等. 生物金属-有机骨架材料中药物吸附及扩散的分子模拟研究[J].
化学学报, 2014, 72: 942-948. LIU Bei, LIAN Yuan-hui, LI Zhi, et al. Molecular simulation of drug adsorption and diffusion in Bio-MOFs[J]. Acta Chim Sinica, 2014, 72: 942-948. |
The attachment energy (AE) models were employed to predict the growth morphology and the main crystal faces of 2, 6-diamino-3, 5-dinitropyridine-1-oxide(ANPyO) in vacuum. The molecular dynamics (MD) simulation was applied to investigate the interaction of ANPyO crystal faces and N, N-dimethylformamide (DMF) solvent, and the growth habit of ANPyO in DMF was predict using the corrected AE model.