




2. 中国工程物理研究院化工材料研究所, 四川 绵阳 621900
2. Institute of Chemical Materials, China Academy of Engineering Physics, Mianyang 621900, China
炸药在生产、使用和储存等过程中可能会受到不同程度的冲击压缩。如果外界的冲击压缩超出炸药本身的力学或化学稳定性极限, 就会引发炸药的分解, 甚至燃烧或爆炸直至爆轰。因此, 有必要从分子和电子微观层次上详细了解炸药的冲击引发机理, 以利于从本质上了解起爆过程的激发和形成机制。这对新型炸药的分子、晶体和材料设计以及高能材料的安全使用, 均可提供有效的指导和帮助。
由于冲击加载炸药是压缩和加热的耦合作用, 其中涉及到非常复杂的多体化学反应和数量巨大的反应中间体, 这使得从原子水平上研究其引发分解机理十分困难。虽然人们已进行大量冲击压缩炸药的实验研究, 然而, 迄今为止实验尚不能实测到皮秒级瞬间的分解起爆过程。最近发展起来的多尺度冲击方法(MSST)[1]与从头算分子动力学(MD)方法的结合, 为模拟冲击加载炸药的引发分解过程提供了可能。
国内外对冲击作用下炸药晶体所发生物理和化学过程的理论研究已有报道。Jaramillo等[2]运用经典MD模拟研究了冲击加载下奥托今(HMX)的非弹性变形。Cawkwell等[3]采用经典MD方法模拟了黑索今(RDX)冲击诱导剪切带的形成过程。Budzien等[4]运用反应性力场MD方法模拟了冲击波在太安(PETN)中的传播, 进而引发其起爆。Bedrov等[5]采用经典MD方法研究了
在半经验量子MD模拟过程中, 核运动势能场是通过紧束缚方法[14]或其它如AM1、PM3和PM6等近似量子化学方法来计算的, 这使得其精度低于从头算MD方法。由此可见, 运用严格的高精度从头算MD方法揭示炸药晶体的冲击引发机理是十分必要的。
本文选择三大类(氮硝基、碳硝基和氧硝基)典型炸药HMX、TATB和PETN为研究对象, 运用从头算MD和MSST相结合方法, 对它们实施冲击加载下的模拟研究。揭示它们的晶体、分子和电子结构随时间的演变细节; 比较它们在冲击压缩下的引发分解机理; 讨论它们的结构和引发分解机理与冲击感度之间的规律联系。
2 模拟方法本研究的模拟采用CPMD[15-16]软件包中的Born-Oppenheimer(BO)MD和MSST相结合方法。CPMD是基于DFT并利用平面波赝势方法进行从头算MD和第一性原理电子结构计算的程序。MSST方法是一种建立在可压缩流的Navier-Stokes方程基础上的模拟方法[1]。该方法已被证明能用解析的状态方程准确地描述通过炸药反应区的热力学态的变化次序[17]。MSST方法能使用较少原子数和较低计算代价使模拟冲击波成为可能。与此同时, 从头算MD能准确计算原子间力, 从而可预测化学反应。离子实和电子间的相互作用通过Martins-Troullier赝势[18]加以描述。采用广义梯度近似下的Perdew-Burke-Ernzerhof (PBE)[19]泛函。平面波截断能取60 Ry。
HMX、TATB和PETN的模拟体系分别用2×1×1、1×1×2和1×1×2。经过初步测试, 当冲击速度取6500 m·

|
图 1 HMX、TATB和PETN超晶胞 Fig.1 Supercells of HMX, TATB, and PETN |
(1) HMX
图 2为冲击加载下HMX超胞分子结构随时间的变化。由模拟结果可见, 在冲击加载下, 超胞中HMX分子上的硝基振动变形。当加载时间达到0.0142 ps时, 两个HMX环上N—O键首先断裂形成氧自由基。同时, 一个HMX环断裂形成链状自由基。当加载时间达到0.0151 ps时, 一个HMX自由基链发生断裂形成两个短自由基。随后,

|
图 2 冲击加载下HMX超胞分子结构随时间的变化 Fig.2 Time dependence of the molecular structure of HMX supercell under shock loading |
(2)
图 3为冲击加载下TATB超胞晶体结构随时间的变化。在冲击波加载下, 超胞中TATB分子上的取代基发生弯曲, 导致TATB环失去平面性。当加载时间达到0.0968 ps时, TATB分子变成椅式构型。当加载时间达到0.101 ps时, 一个TATB分子上取代基

|
图 3 冲击加载TATB超胞晶体结构随时间的变化 Fig.3 Time dependence of the molecular structure of TATB supercell under shock loading |
(3)
图 4为冲击加载下PETN超胞晶体结构随时间的变化。在冲击加载下, 超胞中PETN分子上的硝基发生振动, 导致整个分子发生扭曲而变形。当加载时间达到0.0090 ps时, 分子中第一个C—O键发生断裂形成O—

|
图 4 冲击加载PETN超胞晶体结构随时间的变化 Fig.4 Time dependence of the molecular structure of PETN supercell under shock loading |
虽然由于模拟时间较短, 三种炸药的分解反应尚未完成, 但模拟结果能够反映他们的引发分解过程。在热和撞击等外界作用下, 先前对于HMX、TATB和PETN三类炸药引发分解的机理, 往往基于实验推测[22]和理论预示[23], 认为分子中最弱的N—
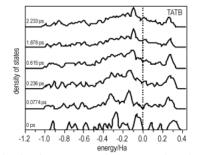
图 5为冲击加载下HMX、TATB和PETN超胞电子结构随时间的变化。由图 5可见, 随着模拟时间的增加, 冲击加载对HMX、TATB和PETN的态密度都产生了显著的影响。顶部价带的态密度峰变得更分散, 表明体系中电子离域性增加; 价带和导带间的带隙逐渐减小; 体系中电荷的重叠不断增加。比较图 5a, 图 5b和图 5c还可看出其不同之处:当加载时间达到0.0217 ps时(图 5a), 在费米能级处,其态密度有一定的数值,表明HMX晶体中出现部分金属态。当加载时间达到0.861 ps时, HMX晶体中出现的较多金属态。这表明随着时间的增加, HMX逐步发生了分解。当加载时间达到0.236 ps时(图 5b), TATB晶体中出现部分金属态。当加载时间达到0.615 ps时, TATB晶体中出现的较多金属态, 表明随着时间的增加, TATB逐步发生了分解。当加载时间达到0.011 ps时(图 5c), PETN晶体中出现部分金属态, 表明随着时间的增加, PETN逐步发生了分解。

|
图 5 冲击加载下HMX, TATB和PETN超胞电子结构随时间的变化(Fermi能级为0) Fig.5 Time dependence of the electronic structure of HMX, TATB, and PETN supercells under shock loading (the Fermi energy is set to zero) |
在冲击波加载下, 随着时间推移, 三种炸药的电子结构产生了类似的变化过程:从态密度图可见, 顶部价带的态密度峰变得更分散, 表明体系中电子离域性增加; 价带和导带间的带隙逐渐减小; 接着开始出现金属态并不断变多, 表明它们开始分解。值得注意的是, PETN、HMX和TATB开始出现金属态的时间依次为0.0107,0.0217,0.236 ps, 与它们感度依次减小的实验事实吻合。
在冲击波加载下, TATB、HMX和PETN晶体引发断裂化学键所需时间分别为0.101,0.0142,0.0090 ps, 其依次减小的顺序, 恰与它们感度(稳定性)依次增大的实验顺序一致, 这进一步证明, 特别注重引发分解步骤对感度即安全性非常重要的理念是正确的。
4 结论本文以从头算MD和MSST相结合的方法模拟冲击加载HMX、TATB和PETN超胞, 展示了它们的分子结构和电子结构在冲击作用下随作用时间推移的细致变化, 着重关注了体系引发分解的化学变化。
HMX超晶胞在冲击加载下, 分子中硝基振动变形, 两个HMX的N—O键优先断裂产生自由基, 同时一个HMX分子发生环断裂形成自由基链; 进而出现
在冲击加载下, TATB分子上取代基首先发生弯曲并变成椅式构型; 接着硝基中C—O键首先断裂生成氧自由基; 继而体系经历六元碳环断裂变成五元环、形成五元稠环、七元环, 进而形成苯并呋咱稠环等网状结构, 并展现它们与自由基共存的奇特图像。值得指出的是, 苯并呋咱稠环是TATB冲击实验中出现的中间产物。
PETN晶体在冲击加载下, 首先发生C—O键断裂; 接着全部C—O键断裂形成
TATB、HMX和PETN晶体在冲击加载下, 引发断裂化学键所需时间分别为0.011,0.0142,0.0090 ps, 其依次减小的顺序, 恰与它们感度(稳定性)依次增大的实验顺序一致。
当然, 从头算MD与MSST相结合方法模拟炸药冲击分解机理的研究刚刚处于起步阶段。许多因素, 如模拟体系的空间尺度、模拟的时间尺度、冲击加载的方向以及速度等等, 还需要作进一步深入细致的研究。
| [1] |
Reed E J, Fried L E, Joannopoulos J D. A method for tractable dynamical studies of single and double shock compression[J].
Phys Rev Lett, 2003, 90: 235503 DOI:10.1103/PhysRevLett.90.235503 |
| [2] |
Jaramillo E, Sewell T D, Strachan A. Atomic-level view of inelastic deformation in a shock loaded molecular crystal[J].
Phys Rev B, 2007, 76: 064112 DOI:10.1103/PhysRevB.76.064112 |
| [3] |
Cawkwell M J, Sewell T D, Zheng L, et al. Shock-induced shear bands in an energetic molecular crystal: Application of shock-front absorbing boundary conditions to molecular dynamics simulations[J].
Phys Rev B, 2008, 78: 014107 DOI:10.1103/PhysRevB.78.014107 |
| [4] |
Budzien J, Thompson A P, Zybin S V. Reactive molecular dynamics simulations of shock through a single crystal of pentaerythritol tetranitrate[J].
J Phys Chem B, 2009, 113: 13142-13151. DOI:10.1021/jp9016695 |
| [5] |
Bedrov D, Hooper J B, Smith G D, et al. Shock-induced transformations in crystalline RDX: A uniaxial constant-stress Hugoniostat molecular dynamics simulation study[J].
J Chem Phys, 2009, 131: 034712 DOI:10.1063/1.3177350 |
| [6] |
He L, Sewell T D, Thompson D L. Molecular dynamics simulations of shock waves in oriented nitromethane single crystals[J].
J Chem Phys, 2011, 134: 124506 DOI:10.1063/1.3561397 |
| [7] |
He L, Sewell T D, Thompson D L. Molecular dynamics simulations of shock waves in oriented nitromethane single crystals: Plane-specific effects[J].
J Chem Phys, 2011, 136: 034501 |
| [8] |
Easom R M, Sewell T D. Shock-induced inelastic deformation in oriented crystalline pentaerythritol tetranitrate[J].
J Phys Chem C, 2012, 116: 2226-2239. DOI:10.1021/jp206826d |
| [9] |
Reed E J, Manaa M R, Fried L E, et al. A transient semimetallic layer in detonating nitromethane[J].
Nat Phys, 2008(4): 72-76. |
| [10] |
Manaa M R, Reed E J, Fried L E, et al. Nitrogen-rich heterocycles as reactivity retardants in shocked insensitive explosives[J].
J Am Chem Soc, 2009, 131: 5483-5487. DOI:10.1021/ja808196e |
| [11] |
Zhu W H, Huang H, Huang H J, et al. Initial chemical events in shocked octahydro-1, 3, 5, 7-tetranitro-1, 3, 5, 7-tetrazocine: A new initiation decomposition mechanism[J].
J Chem Phys, 2012, 136: 044516 DOI:10.1063/1.3679384 |
| [12] |
Ge N-N, Wei Y-K, Ji G-F, et al. Initial decomposition of the condensed-phase β-HMX under shock waves: molecular dynamics simulations[J].
J Phys Chem B, 2012, 116: 13696-13704. DOI:10.1021/jp309120t |
| [13] |
Shan T-R, Wixom R R, Mattsson A E, et al. Atomistic simulation of orientation dependence in shock-induced initiation of pentaerythritol tetranitrate[J].
J Phys Chem B, 2013, 117: 928-936. DOI:10.1021/jp310473h |
| [14] |
Elstner M, Porezag D, Jungnickel G, et al. Self-consistent-charge density-functional tight-binding method for simulations of complex materials properties[J].
Phys Rev B, 1998, 58: 7260-7268. DOI:10.1103/PhysRevB.58.7260 |
| [15] |
Car R, Parrinello M. Unified approach for molecular dynamics and density-functional theory[J].
Phys Rev Lett, 1985, 55: 24712474 |
| [16] |
CPMD code, Version 3.11.1, Copyright IBM Corp 1990-2006[CP], Copyright MPI für FestkKörperforschung, Stuttgart, 1997-2001.
|
| [17] |
Reed E J, Fried L E, Henshaw W D, et al. Analysis of simulation technique for steady shock waves in materials with analytical equations of state[J].
Phys Rev E, 2006, 74: 056706 DOI:10.1103/PhysRevE.74.056706 |
| [18] |
Troullier N, Martins J L. Efficient pseudopotentials for plane-wave calculations[J].
Phys Rev B, 1991, 43: 1993-2006. DOI:10.1103/PhysRevB.43.1993 |
| [19] |
Perdew J P, Burke K, Ernzerhof M. Generalized gradient approximation made simple[J].
Phys Rev Lett, 1996, 77: 3865-3868. DOI:10.1103/PhysRevLett.77.3865 |
| [20] |
Sharma J, Forbes J W, Coffey C S, et al. The physical and chemical nature of sensitization centers left from hot spots caused in triaminotrinitrobenzene by shock or impact[J].
J Phys Chem, 1987, 91: 5139-5144. DOI:10.1021/j100303a053 |
| [21] |
Sharma J, Hoffsommer J C, Glover D J, et al.
In Shock Waves in Condensed Matter[M]. Asay J R, Graham R A, Straub G K, Ed. Elsevier: Amsterdam, 1983: 543 |
| [22] |
Kohler J, Meyer R.
Explosives[M]. VCH, New York, 2007 |
| [23] |
肖鹤鸣.
高能化合物的结构和性质[M]. 北京: 国防工业出版社, 2004.
XIAO He-ming. Structures and Properties of Energetic Compounds[M]. Beijing: National Defense Industry Press, 2004 |
Ab initio molecular dynamics in conjunction with multiscale shock technique was used to study the initial decomposition of HMX, TATB, and PETN crystals under shock wave loading.