




近年来,多硝基咪唑在含能材料领域受到越来越多的关注,如2, 4-二硝基咪唑[1-3],4, 5-二硝基咪唑[4],1-甲基-2, 4, 5-三硝基咪唑[5],1-苦基-2, 4, 5-三硝基咪唑[6]等,其首要原因是:与只含碳的芳香族硝基化合物相比,杂环硝基化合物具有更为有利的氧平衡、密度、以及较好的耐热性[7]。三硝基间氯苯胺是合成耐热炸药的主要中间体。由于氨基的存在,可与化合物中的硝基形成氢键。如果形成分子内氢键,可使其特性密度增大;如果形成分子间氢键,可缩短分子间距,使堆积系数值升高,这均会较大幅度提高化合物的熔点。密度泛函理论(DFT)研究指出氨基硝基咪唑[8]是一种潜在的高能钝感材料,但是其合成方法的报道却很少[9]。本工作旨在探索氨基硝基咪唑及其衍生物的合成、耐热性能,为咪唑类高能钝感耐热炸药的开发应用提供参考。
本课题组以4-硝基咪唑为原料,经硝化、重排、还原,合成出2-氨基-4-硝基咪唑,并与3-氯-2, 4, 6-三硝基苯胺进行缩合,合成出尚未见文献报道的新型含能材料2-(3-氨基-2, 4, 6-三硝基)苯胺基-4-硝基咪唑。
2 实验部分 2.1 试剂与仪器试剂:4-硝基咪唑,工业品;乙酸、乙酸酐、发烟硝酸、氯苯、铁粉等均为分析纯;3-氯-2, 4, 6-三硝基苯胺[10],自制。
仪器:Bruker-Avance DRX 500 MHz核磁共振仪(瑞士);Finnigan TSQ Quantum ultra AM型质谱仪(美国);岛津IRPrestige-21型傅里叶变换红外分光光度计型红外光谱仪;DSC823e差示扫描量热仪(瑞士METTLERTOLED公司)。
2.2 实验原理

|
Scheme 1 |
将4.0 g (35.4 mmol) 4-硝基咪唑加入到7.1 mL AcOH和6.8 mL Ac2O的混合溶液中,室温下缓慢滴加3.0 mL发烟硝酸,滴加完毕室温反应10 h,倒入冰水中,过滤,滤液用二氯甲烷萃取,旋干,与滤饼合并,空气干燥得5.26 g,收率94%。
1H NMR(CDCl3) δ: 8.40(s, 1H, C2—H), 8.53(s, 1H, C5—H)。
2.3.2 2, 4-二硝基咪唑的制备将3.00 g (19 mmol) 1, 4-二硝基咪唑加入到30 mL氯苯中,120 ℃下回流8 h,静置冷却,过滤,水洗,干燥得2.64 g,收率88%。
1H NMR (DMSO-d6) δ: 8.46(s, 1H, C5—H); MS(ESI): 157(M—H)。
2.3.3 2, 4-二硝基咪唑的还原将0.37 g (6.6 mmol) Fe加入到25 mL溶有0.32 g (2.0 mmol) 2, 4-二硝基咪唑的冰乙酸中,室温下搅拌30 min,过滤,滤液倒入冰水中,调pH至弱酸性,乙酸乙酯萃取,无水硫酸钠干燥,旋干得0.14 g,收率54%,m.p. 236.32 ℃(dec.)。
1H NMR(DMSO-d6) δ: 5.96(s, 2H, NH2), 7.75(s, 1H, C5—H), 11.49(s, 1H, N-H); IR(cm-1) : 3477(NH2), 3372(NH2), 3156(C—H), 1633(NH面内弯曲), 1537(NO2), 1444, 1372(NO2), 1298, 1230 1105, 965, 804, 715; MS(ESI): 127(M—H)。
2.3.4 2-(3-氨基-2, 4, 6-三硝基)苯胺基-4-硝基咪唑的合成将0.25 g (2.0 mmol) 2-氨基-4-硝基咪唑溶于10 mL DMF,加入0.1 g (1.8 mmol) KOH,室温搅拌1 h,分批加入0.5 g (1.9 mmol) 3-氯-2, 4, 6-三硝基苯胺,搅拌8 h,倒入冰水中,过滤,干燥得0.32 g粗产品,柱层析(EA/PE=1/1)分离,得0.10 g产品,收率15%,m.p. 244.86 ℃(dec.)。
1H NMR(DMSO-d6) δ: 8.13(s, 1H, Cimid5—H), 8.41(s, 2H, NH2), 8.94(s, 1H, C5—H), 10.46 (br, 1H, NH), 12.79(br, 1H, NH imid); IR(cm-1): 3453(NH2), 3343(NH2), 3146(C—H), 3092(C—H), 1619 (NH面内弯曲), 1575(芳环), 1530(NO2), 1480(芳环), 1441, 1356(NO2), 1270, 1115, 934, 811, 755, 724; MS(ESI): 353(M—H)。
3 结果与讨论 3.1 2, 4-二硝基咪唑的选择性还原用Fe粉进行还原时,还原反应通常发生在电荷密度较低的部位。根据文献[11]报告的2, 4-二硝基咪唑的晶体结构,我们知道2位的NO2与咪唑环共平面,而4位却偏离咪唑环平面。在酸性条件下,咪唑环表现为缺电子离域(N1—C2—N3,Scheme 2),因此,2位硝基的电荷密度比4位的低,优先被还原;而且根据文献[12]报道,1-烷基-2, 4-二硝基咪唑的还原均发生在2位;通过与2-苯胺基-4-硝基咪唑[13]的核磁数据(C5 δ=7.8)对比,可进一步确定2位被还原。

|
Scheme 2 |
根据最终产物,我们推测了2-氨基-4-硝基咪唑与3-氯-苦基胺的两种可能反应历程:一种是:2-氨基-4-硝基咪唑中的氨基直接与3-氯-2, 4, 6-三硝基苯胺进行亲核取代反应,形成缔合物,然后在碱性条件下脱去HCl得到目标化合物(Scheme 3);第二种:首先是2-氨基-4-硝基咪唑在碱性条件下中和掉1位质子,然后经过H重排,与3-氯-2, 4, 6-三硝基苯胺进行亲核取代反应形成缔合物,最后脱去Cl-得到目标化合物(Scheme 4)。

|
Scheme 3 |

|
Scheme 4 |
在样品量分别为1.2100 mg和1.1300 mg,N2流速30 mL·min-1,温度范围为50~500 ℃,升温速率为10.00 ℃·min-1的条件下对合成的两种产物进行DSC分析,其结果如图 1所示。从图 1可以看出,2-氨基-4-硝基咪唑的DSC曲线(虚线)在172.45~260.47 ℃之间有一个大的放热峰,峰值温度为236.32 ℃,曲线积分计算得其分解热为84.11 kJ·mol-1,曲线在260.47~362.04 ℃之间还有一个小的放热峰,峰值温度为315.42 ℃,积分计算得其对应的分解热为31.23 kJ·mol-1;2-(3-氨基-2, 4, 6-三硝基)苯胺基-4-硝基咪唑(实线)的DSC曲线在224.08~262.16 ℃之间有一个尖的放热峰,峰值温度为244.86 ℃,其对应的分解热为154.56 kJ·mol-1,在309.84~384.89 ℃之间有一个宽的放热峰,峰值温度为344.55 ℃,其对应的分解热为148.44 kJ·mol-1。

|
图 1 2-氨基-4-硝基咪唑和2-(3-氨基-2, 4, 6-三硝基)苯胺基-4-硝基咪唑的DSC曲线 Fig.1 DSC curves of 2-amino-4-nitroimidazole and 2-(3-amino-2, 4, 6-trinitro)phenylamino-4-nitroimidazole |
(1) 合成了2-氨基-4-硝基咪唑,经核磁共振、质谱及红外确定了化合物结构;探讨了还原的选择性,结果表明2, 4-二硝基咪唑的2位硝基优先被还原;研究了其热分解,分解温度为236.32 ℃。
(2) 设计合成出2-(3-氨基-2, 4, 6-三硝基)苯胺基-4-硝基咪唑,收率15%,经核磁共振、质谱及红外确定了其结构;研究了其热分解,分解温度为244.86 ℃,具有较好的耐热性。
| [1] |
Damavarpu R, Jayasuriya K, Theodore V, et a1. 2, 4-Dinitmimidazole a less sensitive explosive and propellant made by thermal rearrangement of molten 1, 4-dinitroimidazole: USP 5387297[P], 1995.
|
| [2] |
刘慧君, 樊月琴, 曹端林, 等. 微波辅助合成2, 4-二硝基咪唑[J].
含能材料, 2010, 18(1): 1-3. LIU Hui-jun, FAN Yue-qin, CAO Duan-lin, et al. Synthesis of 2, 4-dinitroimidazole by microwave heating[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2010, 18(1): 1-3. |
| [3] |
刘慧君, 杨林, 曹端林. 由1, 4-DNI热重排制备2, 4-DNI的研究[J].
含能材料, 2005, 13(3): 141-143. LIU Hui-jun, YANG Lin, CAO Duan-lin. Preparation of 2, 4-dinitroimidazole by thermal rearrangement of 1, 4-DNI[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2005, 13(4): 141-143. |
| [4] |
杨国臣, 刘慧君, 曹端林. 4, 5-二硝基咪唑的制备[J].
含能材料, 2006, 14(5): 349-351. YANG Guo-chen, LIU Hui-jun, CAO Duan-lin. Preparation of 4, 5-dinitroimidazole[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2006, 14(5): 349-351. |
| [5] |
Cho J R, Kim K J, Cho S G, et al. Synthesis and characterization of 1-methyl-2, 4, 5-trinitroimidazole(MTNI)[J].
Journal of Heterocyclic Chemistry, 2002, 39(1): 141-147. DOI:10.1002/jhet.v39:1 |
| [6] |
Jadhav H S, Talawar M B, Sivabalan R, et al. Synthesis, characterization and thermolysis studies on new derivatives of 2, 4, 5-trinitroimidazoles: Potential insensitive high energy materials[J].
Journal of Hazardous Materials, 2007, 143: 192-197. DOI:10.1016/j.jhazmat.2006.09.014 |
| [7] |
Duddu R, Dave P R, Damavarapu R, et al. Nucleophilic substitution reactions of 1-methyl-2, 4, 5-trinitroimidazole(MTNI)[J].
Synthetic Communications, 2009, 39(23): 4282-4288. DOI:10.1080/00397910902898635 |
| [8] |
Ravi P, Gore G M, Tewari S P, et al. A DFT study of aminonitroimidazoles[J].
Journal of Molecular Modeling, 2011 |
| [9] |
Duddu R, Dave P R, Damavarapu R, et al. Synthesis of N-amino-and N-nitramino-nitroimidazoles[J].
Tetrahedron Letters, 2010, 51: 399-401. DOI:10.1016/j.tetlet.2009.11.046 |
| [10] |
LU Ming, Lü Chun-xu. Synthesis of 2, 6-bis(2', 4', 6'-trinitro-3'-aminoanilino)-3, 5-dinitropyridine[J].
Huaxue Shijie, 2002, 41(3): 135-138. |
| [11] |
Bracuti A J. Crystal structure of 2, 4-dinitroimidazole(2, 4-DNI)[J].
Journal of Chemical Crystallography, 1995, 25(10): 625-627. DOI:10.1007/BF01665967 |
| [12] |
Olender D, Żwawiak J, Zaprutko L. Selective reduction of 2, 4-dinitro-and 4, 5-dinitroimidazole derivatives using iron dust[J].
Journal of Heterocyclic Chemistry, 2010, 47(5): 1049-1055. DOI:10.1002/jhet.v47:5 |
| [13] |
Sharnin G P, Fassakhov R Kh, Eneikina T A, et al. Studies on five-membered heterocycles. Ⅰ. Synthesis of nitrochloroimidazole[J].
Khimiya Geterotsiklicheskikh Soedinenii, 1977(5): 653-655. |
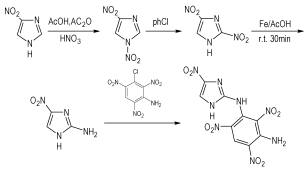
2-Amino-4-nitroimidazole was synthesized using 4-nitroimidazole as starting materials, followed by nitration thermal rearrangement and selective reduction. The condensation of 2-amino-4-nitroimidazole with 3-chloro-2, 4, 6-trinitrobenzenamine led to obtain 2-(3-amino-2, 4, 6-trinitro)phenylamino-4-nitroimidazole.